La protéomique est l'étude de l'ensemble des protéines exprimées dans un organite, une cellule, un tissu, un organe ou un organisme à un temps donné et dans des conditions données. L'ensemble de ces protéines est nommé protéome.
La protéomique permet d'identifier, de caractériser et de quantifier les protéines. L'analyse protéomique est une étude dynamique : un même génome peut conduire à plusieurs protéomes différents en fonction des cellules ou tissus étudiés, des conditions physiologiques ou environnementales, de l'état physiopathologique. Outre leur rôle structural, les protéines ont un rôle fonctionnel et sont impliquées dans de nombreux processus comme le métabolisme, la division cellulaire, la signalisation cellulaire, les échanges avec le milieu extérieur,...
Nous vous proposons d'étudier les protéomes de vos échantillons en appliquant nos techniques préparatives (extraction, concentration, digestion des protéines), séparatives (électrophorèse, chromatographie) et d'analyse (Spectrométrie de masse haute résolution, ELISA).
La protéomique par LC-MS/MS permet de décrire qualitativement (identification des protéines et de leurs modifications post-traductionnelles) et quantitativement les protéomes.
L'analyse génomique permet d'identifier les gènes d'un organisme, mais ne statue pas quant à leur expression (transcription) dans une cellule donnée et dans des conditions données. De même, l'analyse transcriptomique (étude des ARN messagers) permet d'identifier les gènes qui sont exprimés ou transcrits dans une cellule donnée et dans des conditions données, mais ne statue pas quant à la production de protéines fonctionnelles et à la teneur de ces protéines.
Des événements de régulation de la transcription ou de la traduction, comme l'épissage alternatif, ainsi que des modifications post-traductionnelles, comme la phosphorylation, la déamination ou la glycosylation, conduisent pour un même gène à des fonctions différentes. Cette affirmation est soutenue par les résultats des projets de séquençage montrant que les génomes possèdent moins de gènes que de fonctions biologiques décrites.
D'un point de vue quantitatif, la quantification des ARN messagers n'est pas le reflet de la teneur en protéines. En effet, la vitesse de production des protéines ainsi que leur durée de vie sont variables. Par exemple, une protéine dont la synthèse est lente mais la durée de vie longue pourra être présente en plus forte teneur qu'une protéine dont la synthèse est rapide mais la durée de vie brève.
L'analyse en protéomique suppose non seulement des techniques expérimentales, mais également des techniques d'analyse puissantes des données. Notre laboratoire dispose de l'équipement et du savoir-faire nécessaires à leurs mises en oeuvre.
Notre laboratoire met à votre disposition son savoir-faire en matière d'extraction de protéines dans des milieux complexes en définissant un protocole prenant en compte les exigences de rendement, de purification et de préservation de l'intégrité des protéines.
Plusieurs méthodes chromatographiques peuvent être utilisées pour séparer les protéines de vos échantillons selon leur nature et la problématique. Des méthodes électrophorètiques peuvent également être mises en oeuvre pour augmenter la résolution de la technique.
Nous disposons d'un équipement de spectrométrie de masse haute résolution. Cet équipement permet de répondre à toutes les problématiques rencontrées en protéomique : identification, caractérisation et quantification de protéines.
Nous mettons à votre disposition nos outils de bioinformatique pour analyser vos résultats comme par exemple, l'interrogation de bases de données par différents algorithmes pour identifier vos protéines, ou l'utilisation de méthodes d'analyse innovantes telle que le DIA
Nous pouvons appliquer cette démarche à des échantillons divers comme des fluides biologiques, des tissus ou cultures cellulaires d'origine animale ou végétale, des microorganismes ou une simple bande découpée de votre gel d'électrophorèse. Nous avons également une expérience dans le domaine d'étude des sub-protéomes (organites).
Nous sommes à votre écoute pour répondre à vos questions concernant cette démarche et son adéquation avec vos besoins.
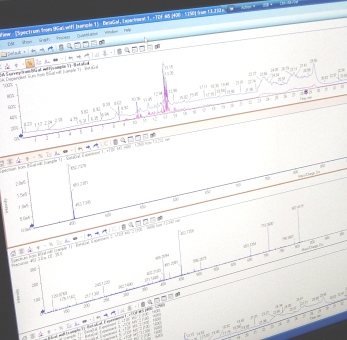
Un instrument de type LC-MS/MS analyse en continu les fractions issues d'une séparation HPLC des peptides issus de la digestion enzymatique d'un extrait protéique. Le spectromètre de masse effectue chaque seconde un spectre MS suivi de 30 MS/MS sur les composés les plus intenses. Ce grand nombre d'informations, générées sur les différents segments des protéines, est comparé à des cartes de masses et spectres MS/MS théoriques de protéines référencées dans des banques de données permettant ainsi d'identifier les protéines présentes dans l'échantillon analysé.
L'efficacité et la fiabilité de la méthode « Shotgun proteomics » repose sur le nombre de peptides analysés et sur la précision de la mesure de la masse de chaque peptide ; l'utilisation d'un spectromètre de masse haute résolution répond à ces besoins.
Pour augmenter le nombre de protéines identifiées, il est possible de réaliser deux séparations chromatographiques successives (chromatographie bidimensionnelle) ou de séparer les protéines par électrophorèse et analyser par LC-MS/MS des fractions du gel d'électrophorèse.
Cette technique est en particulier utilisée pour contrôler les caractéristiques de protéines recombinantes. Des modifications de conditions dans un bioréacteur peuvent changer les patterns de glycosylation, phosphorylation ou la formation de ponts cystéine d'une protéine et ainsi modifier sa masse. La grande précision en masse des instruments de spectrométrie de masse haute résolution (1mDa) permet d'identifier ces variations même faibles.
Cette technique est applicable à des protéines entières purifiées ou en mélange peu complexe. La séparation chromatographique est effectuée sur des colonnes de faible hydrophobicité, la masse de la protéine est déduite de son spectre MS.
Les modifications post-traductionnelles (PTM) sont des modifications chimiques des protéines comme la phosphorylation, la déamination ou la glycosylation, qui modifient, activent ou inhibent leurs fonctions biologiques. Les PTM sont impliquées dans plusieurs phénomènes physiologiques tels que la transmission d'un signal cellulaire ou pathologiques ou l'oncogenèse.
Des étapes d'enrichissement par des méthodes chromatographiques spécifiques appliquées lors des étapes d'extraction ou de séparation peuvent être nécessaires. L'analyse du spectre de fragmentation MS/MS permet de mettre en évidence la localisation des modifications sur la séquence protéique et/ou le taux de modification.
Cette méthode est une méthode de quantification relative permettant de comparer les teneurs en protéines de plusieurs échantillons. Elle peut par exemple être utilisée pour comparer un échantillon « sain » et un échantillon « pathologique », deux conditions de culture, des temps de culture différents, ...
Cette méthode repose sur le marquage des peptides issus de la digestion des échantillons par de la trypsine par le réactif TMT®. Lors de la fragmentation MS/MS, le réactif génère un ion fragment de masse connue. Plusieurs réactifs TMT® de même masse (isobarique) mais générant des ions fragments de masses différentes existent. Ils peuvent être utilisés pour marquer des peptides issus de digestats différents qui peuvent alors être mélangés dans un ratio 1:1 lors d'une même analyse LC-MS/MS. La quantité d'ions fragments issus du marqueur est mesurée, elle reflète celle du peptide marqué et de fait de la protéine dont il est issu. La comparaison des quantités d'ions fragments issus des différents marqueurs utilisés pour les différents échantillons permet la quantification relative des protéines. Ces protéines sont par ailleurs identifiées par comparaison des masses et spectres MS/MS des peptides qui en sont issus avec ceux de protéines décrites dans des bases de données.
Cette méthode permet de comparer qualitativement et quantitativement les protéomes de différents individus. Typiquement cette technique peut être utilisée pour comparer deux états, par exemple sain vs pathologique, et permet ainsi d'identifier des biomarqueurs dont la présence ou la teneur témoigne d'un état physiologique donné.
Elle consiste à comparer les profils LC-MS des échantillons et à en estimer l'aire des pics des différents peptides. Cette aire est rapportée à celle acquise pour les mêmes peptides dans un échantillon de référence et ainsi on peut quantifier relativement la teneur en protéine.
Lors du même essai, en analysant les données acquises par LC-MS/MS il est possible d'identifier les protéines impliquées en comparant les masses et spectres des peptides observés à des bases de données de protéines.
Cette méthode permet de générer un très grand nombre de spectres MS/MS pour un même échantillon et ce pour une gamme de masses très étendue. L'emploi d'un spectromètre de masse haute résolution disposant d'une grande vitesse d'acquisition des données est nécessaire.
Cette technique, en améliorant la reproductibilité et la spécificité du dosage, comparativement aux méthodes de quantification « Label-Free » classiques, permet la quantification absolue des protéines dans un échantillon.
Cette méthode est basée sur la quantification de peptides identifiés au préalable comme étant spécifiques de la protéine dont on souhaite mesurer la teneur. Ces peptides sont les seuls analysés par MS/MS, le bruit de fond de la méthode est réduit, sa spécificité et sa sensibilité sont améliorées.
En ajoutant un standard interne aux échantillons, il est possible de quantifier de manière absolue ces peptides et de fait les protéines dont ils sont issus.
De par sa rapidité et sa robustesse, cette méthode est particulièrement adaptée au screening ou à la quantification de biomarqueurs.
Nos équipes sont à votre écoute pour discuter de votre projet afin de définir les meilleures stratégies expérimentales pour répondre à vos questionnements. formulaire de contact
Phylogene, créée en 1999, est une société de services en biotechnologie utilisant les techniques de génomique, transcriptomique, protéomique, métabolomique et bioinformatique associée pour des applications d’identification et d’authentification ou pour mettre en évidence des effets biologiques.
 Accueil
Accueil La Société
La Société Solutions
Solutions Technologies
Technologies News
News Contact
Contact
